7.4. Seaborn#
Seaborn は、データの可視化に特化したライブラリのひとつで、Matplotlib を補完する役割を担っています。Matplotlib では複雑なコードが必要となるようなグラフも、Seaborn を使えば、シンプルな関数ひとつで手軽に描画できるようになります。また、Seaborn は基本的に Pandas と組み合わせて使用することを想定されており、データの受け渡しは Pandas のデータフレームを通して行うのが一般的です。
7.4.1. Seaborn ライブラリー#
Seaborn ライブラリを利用するには、次のようにインポートします。
import seaborn as sns
なお、Seaborn をインポートすると、Matplotlib の標準設定が Seaborn によって上書きされます。そのため、グラフの色合いや背景などのスタイルが、Seaborn をインポートする前と後で変わる点に注意してください。
7.4.2. データセット#
可視化用にどんぐりのデータセットとタネツケバナの遺伝子発現量データセットを読み込みます。
どんぐりのデータセット(acorns.clean.csv)には、さまざまな種類のどんぐり(樹種)に関する情報が記録されています。各サンプルについて、樹種(tree)、重さ(weight)、高さ(height)、直径(diameter)のデータが記録されています。
# !wget https://py.biopapyrus.jp/data/acorns.clean.csv
acorn_data = pd.read_csv('acorns.clean.csv')
acorn_data.head()
| tree | weight | height | diameter | |
|---|---|---|---|---|
| 0 | kunugi | 5.55 | 2.27 | 1.89 |
| 1 | kunugi | 4.62 | 1.98 | 1.84 |
| 2 | kunugi | 5.05 | 2.08 | 1.90 |
| 3 | kunugi | 5.44 | 2.18 | 1.91 |
| 4 | kunugi | 5.60 | 2.20 | 1.93 |
タネツケバナの遺伝子発現量データ(cinsueta_exp.csv)は、Cardamine insueta の葉を水面に浮かべたあと、経過時間(0〜96時間)ごとの遺伝子発現量の変化を測定したデータです。
# !wget https://py.biopapyrus.jp/data/cinsueta_exp.csv
exp_data = pd.read_csv('cinsueta_exp.csv', index_col=0)
exp_data = exp_data.reset_index()
exp_data.head()
| gene | 0h | 2h | 4h | 8h | 12h | 24h | 48h | 72h | 96h | |
|---|---|---|---|---|---|---|---|---|---|---|
| 0 | CARHR000010 | 419 | 306 | 1935 | 2310 | 2976 | 242 | 543 | 739 | 402 |
| 1 | CARHR000060 | 531 | 149 | 981 | 70 | 123 | 348 | 691 | 366 | 160 |
| 2 | CARHR000090 | 27 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 3 | CARHR000110 | 108 | 30 | 93 | 101 | 127 | 55 | 98 | 99 | 75 |
| 4 | CARHR000120 | 14 | 5 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
7.4.3. 基本グラフ#
Seaborn の機能を利用して、散布図や線グラフなどを描く方法を紹介します。まずは、散布図の例です。Seaborn の描画関数は、少なくとも 3 つの引数を取ります。最初にデータセットとしてデータフレーム型のオブジェクトを渡し、次に x 座標と y 座標に対応する列名を指定します。
例えば、散布図を描くには scatterplot 関数を使い、次のように記述します。
sns.scatterplot(acorn_data, x='height', y='weight')
plt.show()
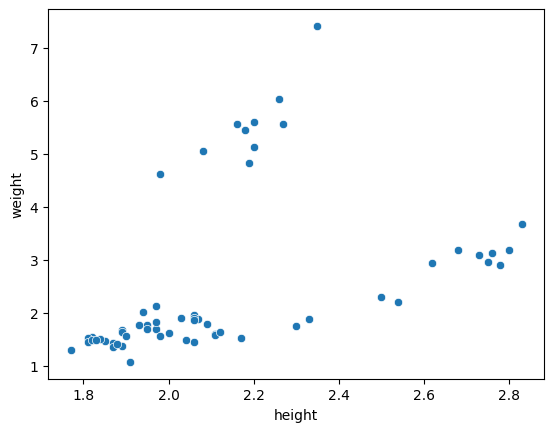
Seaborn の scatterplot 関数では、hue、style、size といったオプションを使うことができます。これらにデータフレームの列名を指定すると、点の色(hue)、マーカーの種類(style)、およびサイズ(size)が、その列に書かれたカテゴリごとに自動的に変わります。
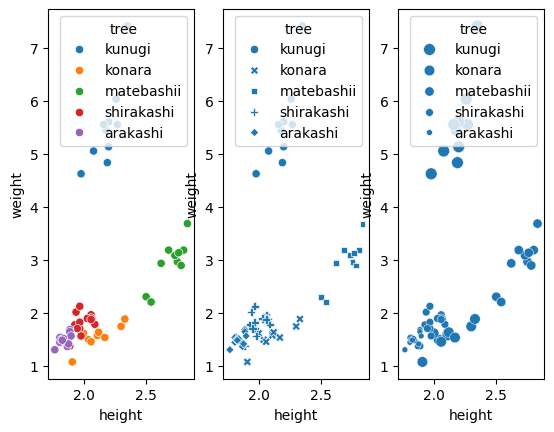
また、scatterplot 関数には ax という引数もあり、ここに fig.add_subplot で作成したサブプロット領域を渡すと、その領域内にグラフを描画できます。では、この機能を使って、hue、style、size の違いを並べて見てみましょう。
fig = plt.figure()
ax1 = fig.add_subplot(1, 3, 1)
ax2 = fig.add_subplot(1, 3, 2)
ax3 = fig.add_subplot(1, 3, 3)
sns.scatterplot(acorn_data, x='height', y='weight', hue='tree', ax=ax1)
sns.scatterplot(acorn_data, x='height', y='weight', style='tree', ax=ax2)
sns.scatterplot(acorn_data, x='height', y='weight', size='tree', ax=ax3)
plt.show()
次に、遺伝子発現量のデータを使って線グラフを描いてみましょう。exp_data はデータフレーム形式で、各行が遺伝子、各列がストレス処理後の経過時間を表しています。Seaborn で遺伝子ごとの発現量の時系列変化を可視化するには、x 軸および y 軸に対応する情報を列名で指定する必要があります。しかし、現在の exp_data にはそれに該当する列がありません。そのため、データフレームを整形し、遺伝子名と経過時間を示す列を追加します。
exp_data 全体で処理すると、情報量が多くなってしまうため、ここでは最初の 5 つの遺伝子のデータのみを対象に処理を行います。また、ここでグラフが見やすいように、発現量を対数化してから可視化します[1]。
exp_data_long = pd.melt(exp_data.iloc[0:5, ], id_vars='gene', var_name='timepoint', value_name='exp')
exp_data_long['log10exp'] = np.log10(exp_data_long['exp'] + 1)
exp_data_long['timepoint'] = exp_data_long['timepoint'].str.replace('h', '').astype(int)
exp_data_long
| gene | timepoint | exp | log10exp | |
|---|---|---|---|---|
| 0 | CARHR000010 | 0 | 419 | 2.623249 |
| 1 | CARHR000060 | 0 | 531 | 2.725912 |
| 2 | CARHR000090 | 0 | 27 | 1.447158 |
| 3 | CARHR000110 | 0 | 108 | 2.037426 |
| 4 | CARHR000120 | 0 | 14 | 1.176091 |
| 5 | CARHR000010 | 2 | 306 | 2.487138 |
| 6 | CARHR000060 | 2 | 149 | 2.176091 |
| 7 | CARHR000090 | 2 | 1 | 0.301030 |
| 8 | CARHR000110 | 2 | 30 | 1.491362 |
| 9 | CARHR000120 | 2 | 5 | 0.778151 |
| 10 | CARHR000010 | 4 | 1935 | 3.286905 |
| 11 | CARHR000060 | 4 | 981 | 2.992111 |
| 12 | CARHR000090 | 4 | 0 | 0.000000 |
| 13 | CARHR000110 | 4 | 93 | 1.973128 |
| 14 | CARHR000120 | 4 | 0 | 0.000000 |
| 15 | CARHR000010 | 8 | 2310 | 3.363800 |
| 16 | CARHR000060 | 8 | 70 | 1.851258 |
| 17 | CARHR000090 | 8 | 0 | 0.000000 |
| 18 | CARHR000110 | 8 | 101 | 2.008600 |
| 19 | CARHR000120 | 8 | 0 | 0.000000 |
| 20 | CARHR000010 | 12 | 2976 | 3.473779 |
| 21 | CARHR000060 | 12 | 123 | 2.093422 |
| 22 | CARHR000090 | 12 | 0 | 0.000000 |
| 23 | CARHR000110 | 12 | 127 | 2.107210 |
| 24 | CARHR000120 | 12 | 0 | 0.000000 |
| 25 | CARHR000010 | 24 | 242 | 2.385606 |
| 26 | CARHR000060 | 24 | 348 | 2.542825 |
| 27 | CARHR000090 | 24 | 0 | 0.000000 |
| 28 | CARHR000110 | 24 | 55 | 1.748188 |
| 29 | CARHR000120 | 24 | 0 | 0.000000 |
| 30 | CARHR000010 | 48 | 543 | 2.735599 |
| 31 | CARHR000060 | 48 | 691 | 2.840106 |
| 32 | CARHR000090 | 48 | 0 | 0.000000 |
| 33 | CARHR000110 | 48 | 98 | 1.995635 |
| 34 | CARHR000120 | 48 | 0 | 0.000000 |
| 35 | CARHR000010 | 72 | 739 | 2.869232 |
| 36 | CARHR000060 | 72 | 366 | 2.564666 |
| 37 | CARHR000090 | 72 | 0 | 0.000000 |
| 38 | CARHR000110 | 72 | 99 | 2.000000 |
| 39 | CARHR000120 | 72 | 1 | 0.301030 |
| 40 | CARHR000010 | 96 | 402 | 2.605305 |
| 41 | CARHR000060 | 96 | 160 | 2.206826 |
| 42 | CARHR000090 | 96 | 0 | 0.000000 |
| 43 | CARHR000110 | 96 | 75 | 1.880814 |
| 44 | CARHR000120 | 96 | 0 | 0.000000 |
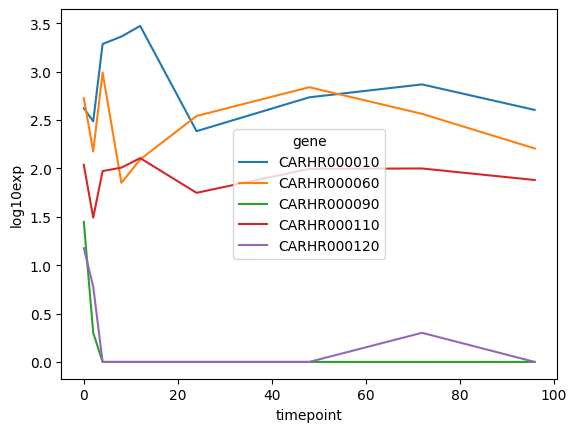
整形後のデータを使い、Seaborn の lineplot 関数で遺伝子ごとの発現量の時間変化を描きます。
sns.lineplot(exp_data_long, x='timepoint', y='log10exp', hue='gene')
plt.show()
7.4.4. ヒートマップ#
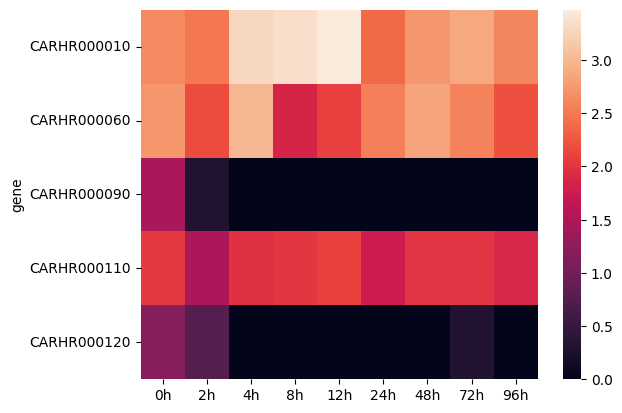
Seaborn の heatmap 関数を使えば、遺伝子発現データのような行列形式のデータから簡単にヒートマップを作成できます。
ここでは、タネツケバナの遺伝子発現量データ exp_data を使い、最初の 5 つの遺伝子の発現量に対してヒートマップを作成します。グラフを見やすくするために、発現量は対数変換(log10)してから可視化します。
expmat = exp_data.iloc[0:5, 1:]
expmat.index = exp_data.iloc[0:5, 0]
expmat = np.log10(expmat + 1)
sns.heatmap(expmat)
plt.show()
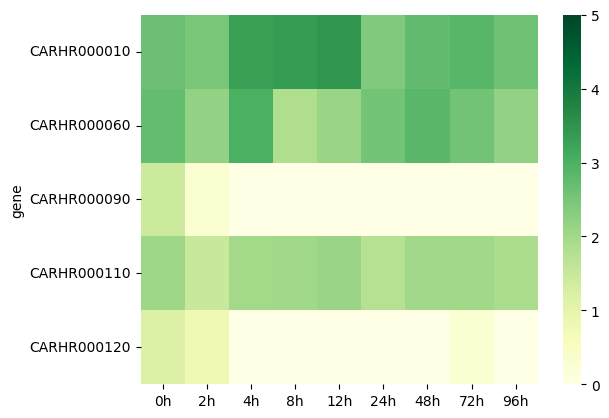
heatmap 関数では、さまざまなオプションを指定することで見た目を調整できます。例えば、vmin および vmax は色スケールの最小値・最大値を指定するのに利用します。また、cmap はカラーパレットを指定するのに利用します。'YlGn'、'viridis'、 'coolwarm' などの Matplotlib で定義されたカラーパレットの名前を指定します。
sns.heatmap(expmat, vmin=0, vmax=5, cmap='YlGn')
plt.show()
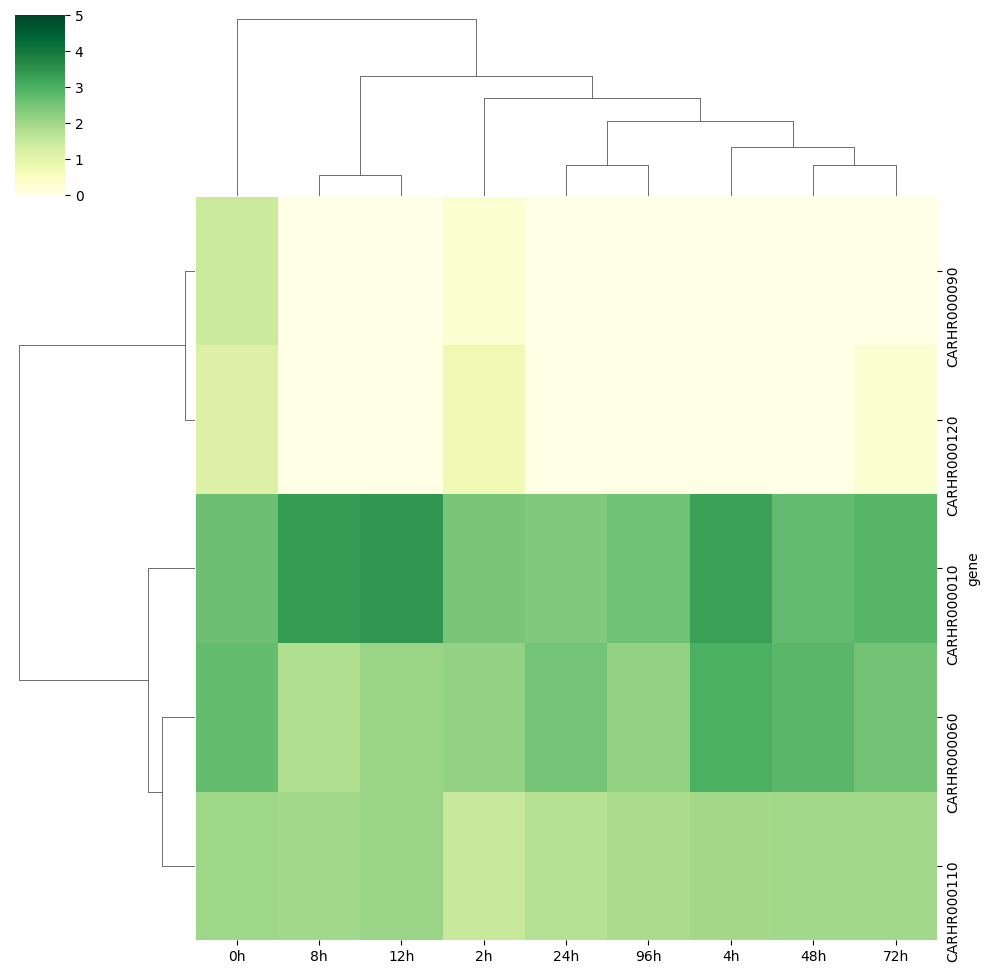
より詳しい分析を行いたい場合は、clustermap 関数を使って、行や列を階層的にクラスタリングしたヒートマップを描くことができます。クラスタリングには、距離の定義('euclidean'、'correlation' など)とクラスタリングアルゴリズム(ward、average など)を指定します。
sns.clustermap(expmat, metric='euclidean', method='ward',
vmin=0, vmax=5, cmap='YlGn')
plt.show()
7.4.5. 多変量データの可視化#
Seaborn には、多変量データの関係性を視覚的に把握できる便利な関数が多数用意されています。ここでは代表的なものをいくつか紹介します。
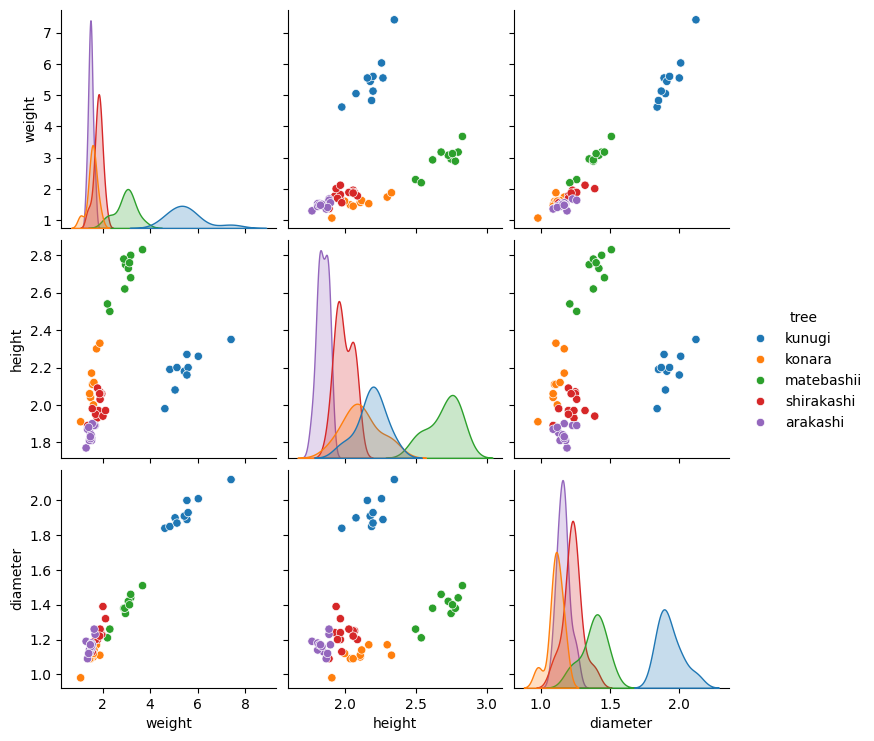
pairplot 関数を使うと、データフレーム内の数値変数の全ての組み合わせについて散布図を自動的に描画してくれます。また、hue オプションを使えばカテゴリごとに色分けも可能です。データ全体の傾向をざっくり把握したいときに便利です。
sns.pairplot(acorn_data, hue='tree')
plt.show()
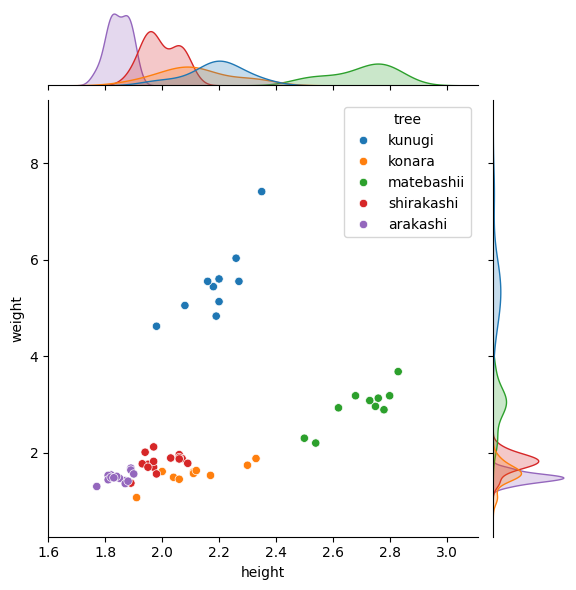
特定の 2 変数間の関係を詳しく見たいときには、jointplot 関数が役立ちます。この関数は、中央に散布図を描き、その周囲に各変数の分布(ヒストグラム)を表示してくれます。
sns.jointplot(acorn_data, x='height', y='weight', hue='tree')
plt.show()
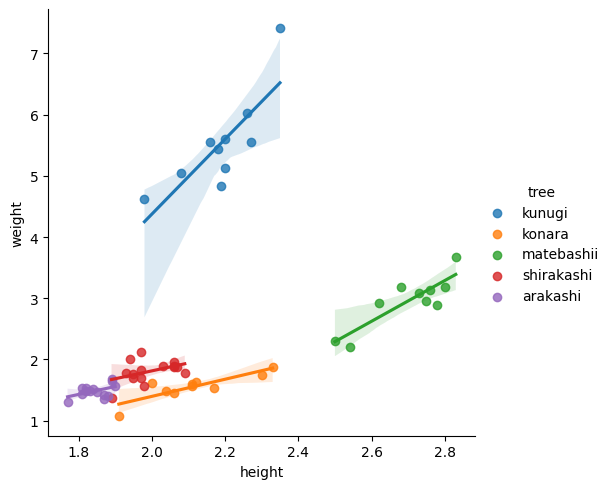
lmplot 関数は、散布図と共に回帰直線を描画し、さらにその 95% 信頼区間も表示してくれます。2 変数間にどのような傾向があるのかを視覚的に確認するのに適しています。
sns.lmplot(acorn_data, x='height', y='weight', hue='tree')
plt.show()
Seaborn では、上記以外にも多彩なグラフを簡単に作成できます。描きたいグラフがある場合は、Seaborn の公式ドキュメントやインターネットで検索して調べると良いでしょう。ドキュメントには例付きの丁寧な説明が掲載されています。
前節および本節では、Matplotlib や Seaborn を使ったさまざまなデータ可視化の例を紹介しました。グラフはデータを「伝える」ための道具です。適切な可視化を選び、見せたい情報がしっかり伝わるよう心がけましょう。で、ここに書いたことを全部覚えようなんて、さすがに思ってませんよね?正直に言うと、たとえ覚えたとしても数週間後にはほとんど忘れてます。それに、数ヶ月後のライブラリのアップデートであっさり消えてしまうかもしれませんからね。